
SMARTINI3 parametrization of multi-scale membrane models via unsupervised learning methods

Force field
In this study, each lipid molecule is characterized by one hydrophilic head and two hydrophobic tails, representing choline and phosphate groups, and the fatty chain of a lipid molecule, respectively. To achieve this representation, we employed a three-site coarse-grained (CG) lipid model, as illustrated in Figure 1A. It is notable that the lipid model utilized in this study is discussed elsewhere in the literature35. The Hamiltonian of the three-site CG model, considering both bonded and nonbonded interactions, is presented below:
$$\beginaligned U=U_bond+U_angle+U_nonbonded, \endaligned$$
(1)
where \(U_bond=0.5k_bond(r-r_0)^2\) and \(U_angle=0.5k_angle(\theta -\theta _0)^2\) denote the harmonic stretching and bending potential, respectively, whereas \(U_nonbonded\) represents the nonbonded potential for intermolecular interactions. Here, \(k_bond\) and \(k_angle\) are the force constants , \(r_0\) is the equilibrium bond length (1nm) , and \(\theta _0\) is the equilibrium angle (\(30^\circ\)). Recent studies in the field40,41,42 have underscored the importance of the attractive potential between lipid tails in implicit solvent models as a crucial factor in replicating the equilibrium structure and self-assembly properties of bilayers. Therefore, the three-site CG model introduced in this study also incorporates an attractive potential between hydrophobic tails. In general, the nonbonded interactions are divided into two components: a repulsive short-ranged interaction, rapidly diminishing with distance, utilized to simulate the excluded volume effect between CG sites, and an attractive long-ranged interaction that incorporates the effective potential of solvent effects. Ultimately, the following combined potential form is employed to characterize the nonbonded interactions among various CG sites:
$$\beginaligned U_nonbonded^TT= & \frac\epsilon 5.5\left[ 0.5 \left( \fracr_minr\right) ^6-6 \left( \fracr_minr\right) ^0.5\right] \endaligned$$
(2)
$$\beginaligned U_nonbonded^HH/HT= & \frac0.4\epsilon 5.50.5 \left( \fracr_minr\right) ^6, \endaligned$$
(3)
where \(\epsilon\) represents the potential well depth and \(r_min\) is the distance of the minimum potential. To mitigate potential artifacts at the cutoff position, the nonbonded potentials are modified using a shift function.
As a result, both the potential and forces exhibit continuity and a smooth decay to zero between the starting position of the shift function, denoted as \(r_1\), and the cutoff distance \(r_c\) (see Figure 1B). The GROMACS shift function used in this work is applied to the force function F(r) and is given by43:
$$\beginaligned S(r,r_1,r_c)=\frac1+A(r-r_1)^2+B(r-r_1)^3r^-(\alpha +1), \endaligned$$
(4)
where \(\alpha\) is the interaction power which is 0.5 and 6 for attraction and repulsion in our three-site CG model , respectively. In addition, the constants A and B are obtained according to the conditions that the function should be smooth at \(r_1\) and \(r_c\) , hence :
$$\beginaligned A= & -\frac(\alpha +4)r_c-(\alpha +1)r_1r_c^\alpha +2(r_c-r_1)^2 \endaligned$$
(5)
$$\beginaligned B= & \frac(\alpha +3)r_c-(\alpha +1)r_1r_c^\alpha +2(r_c-r_1)^3 \endaligned$$
(6)
Therefore, the total force function is obtained as follows:
$$\beginaligned F_s(r,r_1,r_c)=\frac1r^\alpha +1+A(r-r_1)^2+B(r-r_1)^3 \endaligned$$
(7)

(A) The schematic drawing depicts the coarse-grained model of a lipid molecule. The hydrophilic bead, represented by the letter “H,” corresponds to the cholesterol and phosphate groups of the lipid, whereas the two hydrophobic tail beads labeled “T” symbolize the fatty acid chains. (B) Original and shifted potential functions: Intermolecular potential energy versus distance of a pair of beads.
Molecular dynamics simulations
Langevin dynamics (LD) was employed for all simulations with a time step of 0.15 ps, utilizing the molecular dynamics (MD) package GROMACS44. The equation of motion in LD for a system of N particles with masses M is obtained as follows:
$$\beginaligned M\overset..X=-\nabla U^LD-\gamma \overset.X+\sqrt2k_B\gamma TR(t), \endaligned$$
(8)
where U(x) represents the interaction potential of the particles; \(\nabla\) is the gradient operator; \(\overset.X\) and \(\overset..X\) denote velocity and acceleration, respectively; \(\gamma\) is the friction coefficient, and the third term is the noise term with T as the temperature, \(k_B\) Boltzmann’s constant and R(t) as delta-correlated stationary Gaussian process with zero-mean. Periodic boundary conditions were applied across all three dimensions. Steepest descent algorithm was used to minimize the energy of all configurations, succeeded by a 200 ps molecular dynamics (MD) simulation in the canonical (NVT) ensemble to equilibrate the systems at a temperature of 315 K using the Nose-Hoover thermostat. Pressure coupling was implemented using the Berendsen barostat. It is noted that various bilayer properties, including area per lipid, area compressibility, and bending modulus, were measured by imposing zero surface tension in the xy plane with a compressibility of \(1\times 10^-5\) 1/bar. Simultaneously, pressure was maintained at 1 bar in the z plane with a compressibility of 0. A relaxation time of 1 ps was employed for both temperature and pressure coupling.
Computational analysis of membrane properties
Area per lipid
Area per lipid is regarded as a key parameter in the characterization of membranes8. The area per lipid plays a crucial role in fine-tuning force fields for membrane systems, making it an indispensable factor. This suggests that accurately calculating structural properties, including the area per lipid, is essential for the molecular dynamics simulation of membranes45. Furthermore, the average area per lipid is associated with various membrane properties, encompassing acyl chain ordering, molecular packaging, and area compressibility46. Moreover, structural dimensions of bilayers, particularly hydrophobic matching and area per lipid, are pivotal parameters in research domains like in vitro studies of integral membrane proteins or peptides, where the frequent reconstitution of membrane lipids is essential47,48,49,50,51,52. In the context of phospholipid membranes, experimental studies have indicated that the typical range for the area per lipid is reported to be between 0.55 and \(0.75\,\hbox nm^2\)53. As an example, Kucerka et al. employed hybrid electron density models and determined the area per lipid for POPC, the most abundant lipid in animal cell membranes, to be 0.683 ± 0.015 \(\hbox nm^2\)54. Experimentally determining membrane properties can be challenging. Consequently, atomistic and coarse-grained molecular dynamics simulations have been extensively employed as an alternative, robust, and reliable method for calculating various membrane properties. In molecular dynamics simulations conducted under an NPT ensemble, the dimensions of the simulation box may fluctuate due to pressure coupling. Therefore, in this study, the average area per lipid was determined by calculating the size of the simulation box in the xy plane using the following relation55:
$$\beginaligned A_Lipid=\frac\left\langle L_xL_y \right\rangle N_lipids, \endaligned$$
(9)
where \(L_x\) and \(L_y\) represent the membrane plane simulation box dimensions and \(N_lipids\) denotes the number of lipids present in one leaflet of the bilayer. Typically, the time evolution of the average area per lipid during the simulation is monitored and visualized graphically. Indeed, fluctuations around an average value are anticipated for the area per lipid. Upon reaching convergence, the average area per lipid is expected to stabilize, showing minimal changes as the system reaches equilibrium. Indeed, Frigini et al.46 emphasized that the time-dependent behavior of the area per lipid can serve as a valuable criterion for evaluating whether a membrane system has achieved a steady state. They conducted an investigation into the impact of glyphosate in both neutral (GLYP) and charged (GLYP2) forms on a fully hydrated DPPC lipid bilayer. This was accomplished by observing the time evolution of the area per lipid. Simulation results indicated that, for both charged glyphosate and neutral glyphosate at various G:L ratios, the systems reached stability after approximately 5-10 nanoseconds (ns) of simulation time46. In a comparative study among multiple phospholipids, including POPC including POPC(\(\hbox C_42\hbox H_82\hbox NO_8\)P), DOPE(\(\hbox C_41\hbox H_78\hbox NO_8\)P), and POPE (\(\hbox C_39\hbox H_76\hbox NO_8\)P), Zhang et al. have reported that POPC and POPE phospholipids exhibit the highest (0.683 \(\hbox nm^2\)) and lowest (0.588 \(\hbox nm^2\)) area per lipid, respectively56. Angladon et al. conducted coarse-grained molecular dynamics simulations at 310 K and reported that the area per POPC molecule is \(0.676\,\hbox nm^2\), which agrees well with the reference value of \(0.683\,\hbox nm^2\)57. Wang et al. developed an implicit solvent coarse-grained (CG) model for quantitative simulations of POPC bilayers. In the smallest membrane system comprising 72 lipids, equilibrated for about 30 ns in the \(\hbox NP_zz\)AT ensemble, the calculated area per lipid was found to be \(0.683\,\hbox nm^2\). This alignment with the saturated area measured in experiments indicates that the solvent-free CG force field can effectively reproduce the saturated area per lipid obtained from experimental data40. Arnarez et al. utilized the Dry Martini force field to replicate various lipid membrane properties, including the area per lipid, bending modulus, and the coexistence of liquid-ordered and disordered domains. For instance, the area per lipid for a POPC bilayer in Dry Martini (0.64 ± 0.1 \(\hbox nm^2\)) was smaller in comparison to that of the Wet Martini (0.66 ± 0.1 \(\hbox nm^2\)). In contrast, the experimental data ranged from 0.62 to 0.68 \(\hbox nm^2\) and was measured between 297 and 323 K18. Employing a new coarse-grained (CG) model for lipid and surfactant systems, Marrink et al. determined the equilibrium area per lipid for DPPC to be \(0.64\,\hbox nm^2\) at 323 K58. Guo et al. utilized a coarse-grained model for peroxidized phospholipids, built upon the MARTINI lipid force field. They conducted simulations with lipids in a box, considering lateral x and y directions and the transverse z direction, at a temperature of 300 K. As a result, the equilibrium area per lipid for POPC and DOPC phospholipids was measured to be 0.640 ± 0.0008 and 0.669 ± 0.0006 \(\hbox nm^2\), respectively59. Srivastava et al. determined the area per lipid for highly coarse-grained lipid models of DLPC, DOPC, and mixed DOPC/DOPS to be \(0.5763\,\hbox nm^2\), \(0.662\,\hbox nm^2\), and \(0.614\,\hbox nm^2\), respectively. These values are slightly lower than those calculated in atomistic systems, which are \(0.582\,\hbox nm^2\) and \(0.674\,\hbox nm^2\) for DLPC and DOPC, respectively. This discrepancy is attributed to the softer effective potential energy landscape in coarse-grained systems, leading them to exhibit a smaller area per lipid value than corresponding atomistic systems32. Porasso et al.60 employed molecular dynamics simulations to investigate the temporal evolution of various lipid properties, including the surface area per lipid for two lipid bilayer systems, namely DPPC (DiPalmitoylPhosphatidylCholine) and DPPS (DiPalmitoylPhosphatidylSerine). The average area per lipid for DPPC was determined to be 0.663 ± 0.008 \(\hbox nm^2\) in the absence of salt at a temperature of 330 K61,62,63. Furthermore, the area per lipid for the bilayer formed by 288 DPPS in the presence of \(\hbox CaCl_2\) at 0.25N was calculated as 0.522 ± 0.007 \(\hbox nm^2\)60. It is important to highlight that in our evolutionary runs, the goal is to achieve a specific target area per lipid of \(0.68\,\hbox nm^2\), a value commonly reported for the area per lipid of a POPC membrane in both experiments and simulations. Nevertheless, converged solutions within the broader range of 0.55– \(0.77\,\hbox nm^2\) are also taken into consideration, aligning with experimental data obtained for PC membranes, as discussed earlier.
Area compressibility modulus
Area compressibility stands as a crucial elastic characteristic often used to gauge the reliability and efficiency of membrane models. Compressibility, defined as the volume change of a material under stress, applies specifically to the lipid bilayer’s resistance to isotropic area dilation, characterized by an area compressibility modulus, \(K_A\)8. Experimental techniques, such as micropipette aspiration, have reported the area compressibility modulus for POPC bilayers in the range of 208–237 mN/m at \(21\,^\circ \hbox C\)64. Furthermore, alternative experimental values ranging from 180 to 330 mN/m, acquired through Infrared measurements at \(25\,^\circ \hbox C\), have been reported for POPC and SDPC in other studies65. In MD simulations, various methods exist to compute the area compressibility modulus of a bilayer membrane. In this study, area compressibility or the stretching modulus \(K_A\) is calculated through an area fluctuation analysis according to the following equilibrium statistical relation:
$$\beginaligned k_A=\frack_BT4(\left\langle L_x^2 \right\rangle -\left\langle L_x \right\rangle ^2), \endaligned$$
(10)
where \(\left\langle L_x \right\rangle\) and \(\left\langle L_x^2 \right\rangle\) denote the mean value and the quadratic fluctuation of the lateral size of the anisotropic simulation box. Guo et al. computed the stretching modulus for Martini coarse-grained POPC and DOPC bilayers, each comprising 512 lipids, to be 379 ± 21 and 357 ± 19 mN/m, respectively. However, these values were observed to overestimate the accepted experimental reference values. Conversely, in larger systems containing 8192 lipids, better agreement was achieved with compressibilities of 245 ± 42 and 230 ± 27 mN/m59. Additionally, Xu et al. determined the area compressibility modulus of a tensionless coarse-grained bilayer membrane model through the thermal fluctuations of the membrane area, yielding \(\hbox K_A\)= 157.5 mN/m, which was smaller than the experimental data for a DPPC bilayer, i.e., \(\hbox K_A\)= 231 ± 20 mN/m35. Similarly, utilizing fluctuations in the membrane area per lipid, Marrink et al. observed that for a large bilayer patch comprising 6400 lipids, the area compressibility was determined to be \(\hbox K_A\)= 260 ± 40 mN/m which is lower compared to the area compressibility of \(\hbox K_A\)= 400 ± 30 mN/m for a smaller system consisting of 256 lipids. The observed discrepancy was attributed to the contribution of undulatory modes in the large system, leading to a decrease in the area compressibility. The value obtained for the large system aligns well with the experimental value reported for DPPC, which is \(\hbox K_A\)= 231 ± 20 mN/m58. Furthermore, Wang et al. investigated the temperature dependence of the area compressibility by analyzing the standard deviation in the area per lipid. They identified a correlation between the compressibility modulus and the phase of the system. They observed that systems with a compressibility modulus lower than 210 mN/m existed in the liquid phase, whereas those with a compressibility modulus higher than 420 mN/m were in the gel phase66. This discovery suggests that the area compressibility can serve as an indicator of membrane phase transitions. Moreover, as an alternative approach, Saeedimasine et al. calculated the area compressibility modulus by determining the derivative of surface tension with respect to areal strain. Consequently, the calculated area compressibility modulus for Pure-POPC was found to be 188 mN/m at the atomistic level. In contrast, at the coarse-grained level, the values varied between 282 and 297 mN/m for different system sizes67. Finally, it is noteworthy to mention that our evolutionary trials are designed to achieve a specific target area compressibility of 230 mN/m for a POPC membrane. This value falls within the reported range for POPC membrane area compressibility obtained through both experimental and computational techniques. However, we also consider solutions within the broader range of 180–330 mN/m, as discussed earlier, since these values align with experimental data for PC membranes.
Line tension
Line tension, alternatively known as edge energy, refers to the excess free energy arising from the existence of free edges35. The line tension of pores in phospholipid bilayers plays a crucial role in pore-mediated molecular transport techniques. The existence of a pore within the bilayer acts as a gateway for extracellular molecules to enter the cell. This pore facilitates passive molecular transport, enabling molecules to traverse the membrane without requiring additional energy expenditure68. Various methods, encompassing both experimental and computational approaches, have been utilized to measure line tension. In general, line tension or edge tension in diverse membrane compositions has been experimentally measured and observed to fall within the range of 8–42 pN69,70,71,72. For example, Portet et al. introduced a novel technique that allows for the measurement of edge tensions in lipid membranes. This method involves the electroporation of giant unilamellar vesicles and the subsequent analysis of the dynamics of pore closure. To assess the edge tension, the researchers applied this technique to membranes with various compositions, including dioleoylphosphatidylcholine (DOPC) and mixtures of DOPC with cholesterol. As a result, the line tension of DOPC was obtained to be 27.7 ± 2.5 pN and 36.4 ± 1.9 pN for membranes made of DOPC/cholesterol at a 5:1 molar ratio. Additionally, the reported range of line tensions for a phosphatidylcholine (PC) bilayer membrane typically falls between 6.5 and 30 pN73. In this study, molecular dynamics simulations are employed to calculate the line tension \(\gamma _L\) from the anisotropy of the pressure within the simulation box. Specifically, this calculation is performed for a bilayer membrane with two free edges in the x-direction, while the membrane is periodically extended in the y-direction (Figure 2)74:
$$\beginaligned \gamma _L=-\fracL_xL_z2 \left( P_yy-\fracP_xx+P_zz2\right), \endaligned$$
(11)
where \(P_xx\) and \(P_yy\) represent the tangential and \(P_zz\) denotes the normal component of the diagonal pressure tensor, \(L_x\) and \(L_z\) are the simulation box dimensions and the prefactor \(\frac12\) is included to account for the fact that the system has two free edges. Employing a similar methodology, Miyazaki et al. obtained the line tension of a pure ribbon-like POPC bilayer membrane to be 35.1 pN75. Similarly, Y. Jiang et al. utilized pressure anisotropy in the simulation box and calculated the line tension within the range of 10–30 pN for the edge of a pure dimyristoylphosphatidylcholine (DMPC) membrane76. Alternatively, employing an implicit solvent coarse-grained (CG) model for POPC bilayers, Wang et al. utilized a straightforward formula, i.e, \(\gamma _L=-\frac12\left\langle \sigma _xx \right\rangle L_yL_z\) to compute the line tension, where \(\sigma _xx\) represents the xx-component of the stress tensor, \(L_y\) and \(L_z\) represent the length of the simulation box in y and z dimensions, respectively. As a result, the calculated line tension value (\(\gamma _L\)= 29 ± 12 pN) fell well within the range of both experimental and atomistic simulation reference values for PC membranes40. Given the scarcity of reliable statistical data concerning the experimentally determined value of the POPC membrane line tension, as highlighted in literature77, our evolutionary runs are aimed at achieving a specific target line tension of 10 pN. This value aligns with the experimental range of PC membrane line tension, typically reported between 6.5 and 30 pN.

Simulation setup for calculation of line tension in a membrane consisting of 376 lipids. (A) Side view. (B) Top view.
Bending modulus
The membrane’s bending modulus plays a crucial role in regulating the out-of-plane bending deformation of a membrane as described in the Canham-Helfrich elastic model59. Bending modulus values were obtained through experimental measurements. Specifically, Rawicz et al. utilized the micropipette vesicle aspiration technique to measure the bending modulus of fluid phase phosphatidylcholine (PC) membranes, i.e, POPC and DOPC, resulting in values of approximately 9.0 \(\times 10^-20\)J and 8.5 \(\times 10^-20\)J, equivalent to around 20 \(\hbox k_B\)T and 19 \(\hbox k_B\)T, respectively64. In computer simulations, as well as in the present study, the bending modulus for lipid bilayers is commonly determined by computing the membrane undulation spectrum. While the theoretical concept of using the membrane undulation spectrum is straightforward, its practical application is highly sensitive to errors. Moreover, it is only applicable in the “long-wavelength” limit and exhibits slow convergence, as noted elsewhere78. Utilizing a Helfrich-type continuum model, the bilayer can be represented as a single mathematical surface, denoted as u(x, y), with a minimum energy state. This model facilitates the characterization of the bilayer. Consequently, the Hamiltonian for thermally excited energy fluctuations takes the following form79:
$$\beginaligned H\ u(x,y) \=\frac12\int \int (K_c|\nabla ^2u(x,y)|^2 + \gamma |\nabla u(x,y)|^2)dxdy, \endaligned$$
(12)
where \(k_c\) and \(\gamma\) represent bending modulus and surface tension, respectively.
The membrane fluctuations are analyzed by employing a Fourier decomposition of the surface:
$$\beginaligned u(\textbf r)=\sum _\textbf q u(\textbf q)e^i\textbf q.\textbf r, \endaligned$$
(13)
where r=(x,y) is a two-dimensional real space vector and q=(\(q_x\),\(q_y\)) being the two-dimensional reciprocal space vector. Due to the quadratic nature of energy in the amplitudes (Eq.12), the equipartition theorem is applied, stating that each Fourier mode has an average energy of \(\hbox k_B\)T/280. In the absence of any in-plane tensions (\(\gamma\)=0), thermal fluctuations as a function of wave number q are expressed by the following equation81:
$$\beginaligned \langle |u_q|^2\rangle =\frack_BTL^2k_cq^4 \endaligned$$
(14)
,where \(\langle |u_q|^2\rangle\) represents the ensemble average of the energy of the undulatory modes, \(k_c\) denotes the bending modulus , \(k_B\) is the Boltzmann’s constant, T is the temperature and L is the periodic simulation box side. If the membrane model accurately replicates the continuum behavior dictated by the Helfrich energy density, a deviation from the \(\hbox q^-4\) behavior is anticipated at short wavelengths. This divergence arises because the continuum behavior is only applicable to high-wavelength undulations1. Various methods can be utilized to extract the height spectrum from simulations of bilayer membranes. These methods enable the extraction of the bending modulus, with the Real Space (RS) method being a commonly employed approach for this purpose. In the Real Space (RS) method, the membrane height field, represented as u(x, y), is interpolated onto a uniform grid of nodes in a two-dimensional (N\(\times\)N) grid (see Figure 3). To mitigate technical complications, such as “empty” grid cells, it is advisable to select a larger value for N in the Real Space (RS) method. Increasing the size of the grid enhances the accuracy of the interpolation and improves the resolution of the height-height spectrum. Moreover, to map the membrane position onto the defined grid, a straightforward approach involves selecting a reference position in each lipid, such as any of the particles or beads belonging to the lipid or the average position of the phosphocholine head group. The height of the reference position for all lipids within a given grid point is then computed, and the average value of the heights is assigned to that grid point. Another approach to mapping lipid molecules onto a uniform grid, without the need for a reference point, involves directly mapping every microscopic degree of freedom. This entails mapping each individual heavy atom or coarse-grained bead to its nearest grid point, irrespective of the lipid to which it belongs. Finally, to acquire the mean power spectrum, the power spectra of all bilayer configurations are averaged. Indeed, numerous publications in the literature have employed the computational approach described above to calculate the bending modulus of membranes. Guo et al. conducted calculations of the bending modulus utilizing a coarse-grained model specifically developed for peroxidized phospholipids, built upon the MARTINI lipid representation. Their findings revealed bending modulus values of approximately 30.0 and 28.3 \hbox k_BT for POPC and DOPC, respectively59. Yuan et al. employed a one-particle-thick fluid membrane model, wherein each particle represented a cluster of lipid molecules, to derive the bending rigidity of membranes. Their computations resulted in bending rigidity values falling within the experimental range of 12–40 \(\hbox k_B\)T22. Srivastava et al. estimated the bending modulus for highly coarse-grained DLPC membranes to be 6.65\(\times\)10\(^-20\)J, approximately equivalent to 15 \(\hbox k_B\)T. Similarly, the bending modulus for a DOPC model was found to be (7.64 ± 0.671)\(\times 10^-20\)J, corresponding to 18-19 \(\hbox k_B\)T32. Chacon et al. calculated a bending modulus of 21 \(\hbox k_B\)T for POPC membranes at 320 K using coupled undulatory (CU) modes to analyze membrane fluctuations. This approach yielded excellent estimates of the bending modulus in line with proposed theoretical predictions82. In a distinct study by Xu et al., a bending modulus value of 4.35\(\times 10^-20\) J, roughly equivalent to 10 \(\hbox k_B\)T, was reported for a coarse-grained DPPC bilayer35. In conclusion, our evolutionary runs aim to reach a specific target bending rigidity of 12 \(\hbox k_B\)T for a pure POPC membrane, as measured in experiments using techniques like electro-deformation or Fluctuation analysis83. Additionally, solutions converging to experimentally observed bending rigidity values between 8 and 42 \(\hbox k_B\)T are deemed valid and fall within an acceptable range.

The schematic depicts the height function in a bilayer membrane, u(x, y), fitted to bead positions observed in selected frames throughout the simulation trajectory.
Phase transition temperature
Considerable research efforts have been directed toward investigating the phase transition in biological membranes, with a particular focus on the ordered (gel) and disordered (liquid-crystalline) lipid phases. These phases play a crucial role in understanding the overall dynamics and structural properties of membranes, given their direct association with the individual lipid components84. Numerous experimental studies have explored the phase behavior of phospholipids, employing a variety of techniques. Suurkuusk et al. conducted a comprehensive investigation using calorimetric and fluorescent studies on multi-lamellar vesicles of DPPC. In their study, they observed a heating scan of multi-lamellar vesicles that exhibited a phase transition process reminiscent of a first-order transition, a finding subsequently confirmed through fluorescent measurements85. Additionally, Davis et al. employed deuterium magnetic resonance to examine the liquid-crystalline, gel phase, and phase transition of multi-lamellar labeled-DPPC vesicles. Their quantitative measurements based on deuterium spectra revealed a sharp decline in moment values as the vesicles were heated, indicating a first-order transition86. In addition to experimental approaches, computational methods have played a significant role in investigating the phase transition in lipid bilayer membranes. For example, the phase transition temperature was determined by observing a sharp rise in enthalpy during the transition from the gel to liquid crystalline states87. Furthermore, efforts were made to address the issue of substantial hysteresis, which can pose challenges in accurately determining the phase transition temperature for the liquid-gel transition. To overcome this challenge, simulations were conducted on a system comprising two distinct halves, with one half in the gel phase and the other in the liquid-disordered phase (refer to Figure 4). This configuration effectively diminishes or eliminates the nucleation barrier necessary to initiate either the liquid-disordered or gel phase, leading to a considerable reduction in hysteresis88. On the other hand, de Vries et al. conducted an atomistic molecular dynamics study to investigate the phase behavior of lecithin (DPPC) in water. Their simulation results revealed the spontaneous formation of a ripple gel phase when a fully hydrated bilayer was cooled below the phase transition temperature7. Accordingly, Leekumjorn et al. employed atomistic molecular dynamics simulations to thoroughly examine the structural properties of saturated DPPC and DPPE lipid bilayers in the vicinity of the main phase transition. Despite their similar chemical structures, with the only difference in their choline and ethanolamine groups, DPPC and DPPE exhibited contrasting transformation mechanisms from an ordered (gel) to a disordered (liquid-crystalline) state. Leekumjorn et al. used the area per lipid and bilayer thickness as criteria to determine the first-order phase transition temperature of DPPC and DPPE bilayers through their simulations. According to their findings, the transition temperature was approximately 305 K for DPPC and 320 K for DPPE. These values were compared to experimental measurements reporting transition temperatures of 315 K for DPPC and 337 K for DPPE. Additionally, using molecular dynamics with Alchemical Steps, Fathizadeh et al. calculated the phase transition temperature from the gel to fluid phases in pure DPPC bilayers, obtaining values within the range of 303.15–323.15 K84. Coarse-grained approaches have proven effective in determining the phase transition temperature of lipid bilayers. Marrink et al. conducted simulations using a coarse-grained (CG) model to investigate the transformation between a gel and a fluid phase in DPPC bilayers. They employed a cooling technique on bilayer patches and proposed a four-stage reversible process consisting of nucleation, growth, limited growth, and optimization. Extrapolating their findings to a macroscopic bilayer, they obtained a transition temperature of 295 ± 5 K, which was in semi-quantitative agreement with the experimental phase transition temperature value for DPPC (315 K)89. Kociurzynski et al. determined the phase transition temperature of pure DPPC by observing a sharp increase in the area per lipid during the transition from the gel to liquid crystalline states. Their simulation results indicated a transition temperature of approximately 305 K, showing a slight discrepancy of about 10 K compared to the experimental value of 315 K for DPPC. This difference suggested a small deviation between the simulated and experimental transition temperatures89,90. Mirzoev et al. employed a systematic structure-based coarse-graining approach with a grained DMPC lipid model to examine the state point dependence of lipid bilayers. Their simulations covered a temperature range of 200–350 K in 25 K intervals. Through the analysis of the area per lipid and lateral diffusion coefficient, they observed a clear transition between 225 and 250 K, signifying a gel-liquid phase transition91. Eventually, Wang et al. utilized coarse-grained solvent-free membrane models to explore the phase transformation in DOPC and POPC membranes. Their simulations indicated that the phase transition in a DOPC membrane occurs within the temperature range of 265–295 K, while in a POPC membrane, it takes place between 280 and 310 K. The values obtained from the simulations were slightly lower than the experimental transition temperatures reported for DOPC (around 253 K) and POPC (around 270 K)73,92,93. As a result, our evolutionary runs aim to achieve a phase transition temperature of 270 K for a POPC membrane, aligning with experimental observations. Additionally, we consider solutions converging toward the experimentally observed phase transition temperature for a PC membrane, spanning from 250 to 315 K.

Phase transition temperature calculation. (A) The simulation snapshot depicts the initial configuration of a bilayer, with one half of the lipids in the gel phase (left) and the other half (right) in the fluid phase. (B) Enthalpy versus time at different temperatures ranging from 100 to 400k. A sudden increase in enthalpy within the temperature range from 240 to 260 K is indicative of a phase transition from the gel to the liquid-disordered phase. The phase transition temperature is then assigned to the middle of the identified phase transition zone, i.e, 250 k.
Radial distribution functions
Radial distribution functions (RDF) are crucial in the analysis of biological membrane systems using molecular dynamics (MD) simulations, offering valuable insights into the system’s short and medium-range structural information94. In this work, the Radial Distribution Function (RDF) is defined according to the formulation by Hub et al.95:
$$\beginaligned g(r)=\fracn(r,r+\Delta r)\rho \Delta V_2D \endaligned$$
(15)
,where r is the distance in the lateral direction, \(\Delta V_2D=2\pi r \Delta r\) being the volume of a shell with radius r and thickness \(\Delta r\) and n(r,r+\(\Delta r\)) is the number of the sites within this shell. Given the significant level of coarse-graining in this study, a comparison was exclusively carried out using the Earth Mover’s Distance (EMD) technique for the tail-tail radial distribution function (RDF). The comparison involved our three-site coarse-grained (CG) model and the tail-tail RDF derived from an atomistic molecular dynamics simulation of a POPC membrane. The EMD method quantifies the minimal cost required to transform one distribution into another96. Hence, if the two RDF distributions-the normalized RDF related to the coarse-grained model and the atomistic RDF-are identical, the resulting EMD value will be 0, signifying their complete similarity.
Density profile
The distribution of mass along the z-axis of the membrane is visualized by computing the mass density profile across the bilayer8. The density profile can offer insights into various properties of the membrane, including the distribution of molecules and the presence of structural features97. In calculating the mass density profile, it is crucial to account for potential fluctuations in the center of mass (COM) of the membrane during the simulation. Hence, in each frame, the coordinates (x, y, z) of all atoms are determined relative to the instantaneous center of mass (COM), with the z-coordinate set to 0. This approach enables an accurate assessment of the distribution of mass along the membrane’s z-axis. Similar to the radial distribution function (RDF), we conducted an Earth Mover’s Distance (EMD)-based comparison of the mass density profile between the head groups in our three-site coarse-grained (CG) model and the atomistic model of a POPC membrane. In this context, if the normalized density profile of the coarse-grained model perfectly matches that of the atomistic density profile, the Earth Mover’s Distance (EMD) value becomes zero.
Genetic algorithms (GA)
Genetic Algorithms (GA) are developed based on genetic laws and Darwin’s theory of evolution aimed at enhancing the optimization of problems with numerous variables98. For example, they are utilized in the determination of molecular docking within protein-DNA complexes or in the generation of new pharmacophore models through electron density topology analysis99,100. Generally, a Genetic Algorithm (GA) typically consists of five distinct phases, as outlined below:
Initial population
Within a Genetic Algorithm (GA), the search space is represented by a population of individuals, where each individual serves as a potential solution to a specific problem. The individuals in a Genetic Algorithm (GA) are encoded as genomes, which are vectors containing a predefined number of parameters. During the initialization phase, each genome is assigned random values within a predetermined range. The random initialization ensures diversity within the initial population, facilitating a broader exploration of the search space. Subsequently, a cost assessment is carried out to evaluate the quality of each genome. This assessment entails comparing the outcomes generated from molecular dynamics (MD) simulations, employing the parameters encoded in the genome, with the target values from a reference database. The cost score is then determined by assessing the degree of agreement between the molecular dynamics (MD) simulation results and the specified target values. Genomes that do not meet predefined criteria or fail to attain a satisfactory cost score are deemed unfit for further consideration. The eliminated unfit genomes are replaced with new genomes in the subsequent generations, maintaining the population size. In Genetic Algorithms (GA), the initial population size is often chosen to be an even number101.
Selection
The selection operator in a Genetic Algorithm (GA) plays a crucial role in exploiting the advantageous traits of the fittest candidate genomes, aiming to improve and enhance them across successive generations. This accelerates the GA’s convergence towards a preferred solution for the optimization problem under study102. Numerous selection methods have been reported in the literature, among which are roulette wheel selection (RWS), tournament selection (TOS), and linear rank selection (LRS). In this study, roulette wheel selection (RWS) is utilized, due to its widespread adoption as a method in Genetic Algorithms (GA) for selecting genomes for the succeeding generation. The roulette wheel selection (RWS) emulates the concept of survival of the fittest. This method efficiently favors genomes with lower cost scores, providing them with a greater probability of being chosen for the next generation103. Specifically, the RWS assigns a probability of selection \(\hbox P_i\) to each genome i based on its cost value. The probability \(\hbox P_i\) for each genome is defined as:
$$\beginaligned P_i=\fracF_i\sum _j=1^PopSizeF_j, \endaligned$$
(16)
where \(\hbox F_i\) corresponds to the cost of the ith genome. Then, a series of N random numbers are generated and compared with the cumulative probability \(C_i=\sum _j=1^iP_i\) of the population. Ultimately, the fit genome i is chosen and transferred into the new population provided that \(C_i-1< U(0,1) \le C_i\), where U(0, 1) is a uniform random number generated between 0 and 1. In RWS, genomes are allocated to consecutive segments on a line, where the size of each genome’s segment is proportionate to its fitness level. Following the generation of a random number, the genome to be selected is determined by identifying the segment on the line that corresponds to the generated random number. The described process continues until the desired number of genomes, often referred to as the mating population, is achieved. This method shares similarities with a roulette wheel, as the size of each segment is directly proportional to its fitness.
Crossover
Undoubtedly, crossover stands out as one of the most crucial operators in Genetic Algorithms (GA), playing a key role in generating offspring that will constitute subsequent generations. The primary objective of the crossover operation in GA is to broaden the exploration of the search space, thereby aiding in the generation of enhanced genomes in future generations104. The standard crossover operator involves taking two parent genomes and producing two offspring genomes. The underlying concept behind this operation is that the offspring genomes have the potential to display superior characteristics compared to their parents by inheriting the best traits from each parent. This blending of genetic material from both parents enables the exploration of new genetic combinations that may contribute to improved solutions in the evolutionary process. In this study, single-point crossover is utilized, given its widespread acceptance as one of the most commonly employed crossover operations. In single-point crossover, the two parental genomes are divided into two segments at a randomly determined crossover point. A new child genotype is then generated by exchanging the second segment of the first parent with the second segment of the second parent105. Figure 5 illustrates the single-point crossover (SPC) operation.
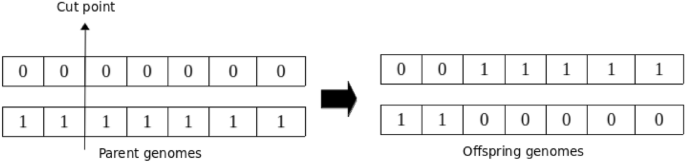
Schematic illustration of single-point crossover. The arrow indicates the crossover cut point.
Mutation
The mutation operator holds significant importance within a Genetic Algorithm (GA) as it plays a crucial role in preventing premature convergence by introducing a random element in the population’s evolution106. The mutation operator is applied to the genomes generated through the crossover operation and involves altering one or more bits within a specific genome, with a probability denoted as \(\hbox P_m\epsilon\)[0,1), referred to as the mutation probability. Hence, the mutation probability governs the frequency at which the bits within a particular genome undergo mutation. Significantly, the mutation probability is adjusted based on the behavior of the population. When the population becomes trapped in a local optimum, the mutation probability is increased. Conversely, when the population is widely spread across the solution space, the mutation probability is decreased. However, it is usually chosen within the range of 0.001–0.05107. Various mutation operators have been proposed for different solution representations, including Gaussian mutation, random mutation, and polynomial mutation108,109,110. In this study, Gaussian mutation is employed as the selected mutation operator, given its widespread recognition as one of the most commonly used mutation operators. Accordingly, for each bit in a given genome, a random value is sampled from a normal distribution with a mean of 0 and a standard deviation of 1. Subsequently, the randomly generated value is compared to a predefined threshold, typically set to 0.05. If the generated value is lower than the threshold, the mutation is considered valid and accepted. However, if the value exceeds the threshold, the mutation is rejected, and no modifications are made to the corresponding bit position within the genome. Once a mutation is accepted, an adaptive mutation operation is executed to introduce additional variability. This operation involves multiplying a sigma value, representing the standard deviation derived from the Gaussian distribution of the bits at a particular position from the previous generation, by the randomly generated number.The resulting value obtained from the adaptive mutation operation is then added to the selected bit within the genome, as illustrated below111:
$$\beginaligned x_i \epsilon \mathbb R \longrightarrow x_i +\sigma N(0,1), \endaligned$$
(17)
where i represents the ith bit of genome and \(\sigma\) denotes the previously discussed standard deviation.
Termination
At this stage of the procedure, the average cost of the best genomes corresponding to a given generation is compared to a threshold, i.e., the target average cost. If the obtained average cost is below the threshold (in the case of a minimization problem) or above the threshold (for a maximization problem), the Genetic Algorithm (GA) terminates. Otherwise, the loop continues until a subsequent generation successfully achieves the goal. It is worth noting that there are alternative stopping criteria for the Genetic Algorithm (GA). These criteria include setting an upper limit on the number of generations or the number of cost function evaluations. Additionally, the Genetic Algorithm (GA) may terminate if the probability of achieving significant changes in the subsequent generation becomes exceedingly low, as mentioned in Ref.112.
Cost function
In this study, the weighted sum method, widely recognized as one of the most prominent approaches for addressing multi-objective optimization problems, is employed to calculate the cost of each genome113. This method involves consolidating multiple objectives into a single aggregated objective function by assigning a weighting coefficient to each respective objective function. Hence, the cost corresponding to each genome is calculated using the following equations:
$$\beginaligned Cost= & Cost_1+Cost_2 \endaligned$$
(18)
$$\beginaligned Cost_1= & \sum _i=1^5w_1,i*\left[ \frac(Simulated\_Property)_i(Target\_Property)_i-1\right] ^2 \endaligned$$
(19)
$$\beginaligned Cost_2= & \sum _i=6^7w_2,i*EMD\left[ (CG\_Simulated\_Property)_i,(Atomistic\_Target\_Property)_i\right] \endaligned$$
(20)
,where \(w_i\) represents the selected weighting coefficient for the ith objective function. Therefore, equation (19) calculates the cost value for five membrane properties, i.e., area per lipid, area compressibility, line tension, bending modulus, and phase transition temperature, each represented by a single value. In contrast, equation (20) is utilized to assess the cost value for the other two properties including radial distribution function and density profile, represented by a distribution. Furthermore, it is important to highlight that the weighting coefficient for each property in the first category is set at 0.15, while for the second category, the assigned weighting coefficient for each property is 0.125. Figure 6 depicts the flow of the genetic algorithm.

Flowchart of the genetic algorithm.
link



